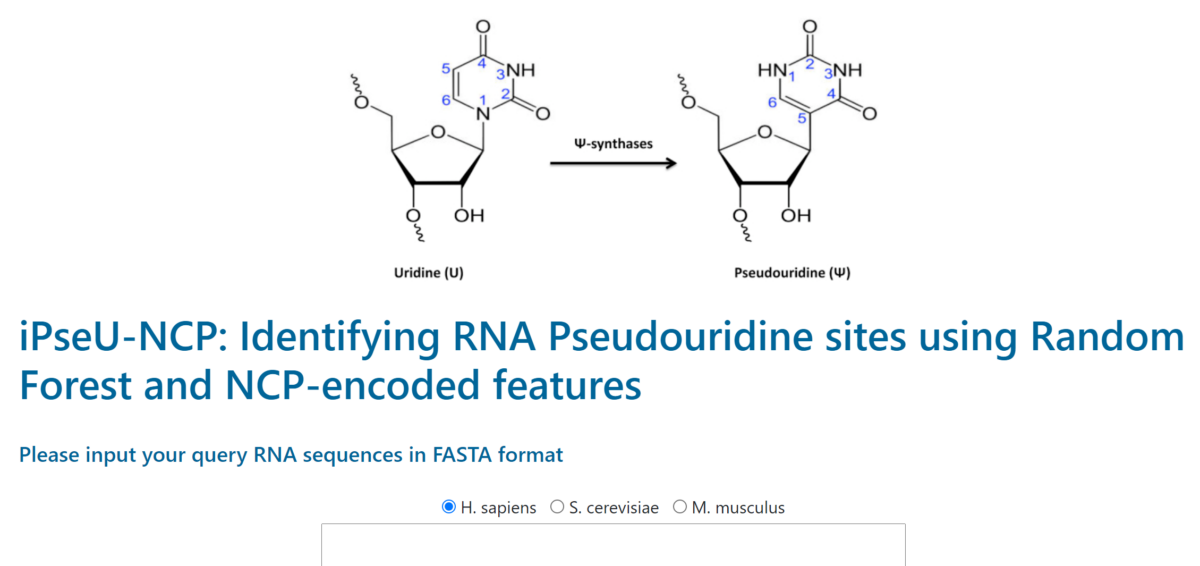
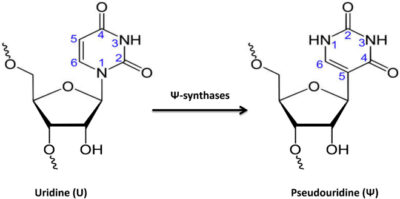
Pseudouridine modification is most commonly found among various kinds of RNA modification that occurred in both prokaryotes and eukaryotes. This biochemical event has been proved to occur in multiple types of RNAs, including rRNA, mRNA, tRNA, and nuclear/nucleolar RNA. Hence, gaining a holistic understanding of pseudouridine modification can contribute to the development of drug discovery and gene therapies. Although some laboratory techniques have come up with moderately good outcomes in pseudouridine identification, they are costly and required skilled work experience. We propose iPseU-NCP – an efficient computational framework to predict pseudouridine sites using the Random Forest (RF) algorithm combined with nucleotide chemical properties (NCP) generated from RNA sequences. The benchmark dataset collected from Chen et al. (2016) was used to develop iPseU-NCP and fairly compare its performances with other methods.
Results: Under the same experimental settings, compared with three state-of-the-art methods including iPseU-CNN, PseUI, and iRNA-PseU, the Matthew’s correlation coefficient (MCC) of our model increased by about 20.0%, 55.0%, and 109.0% when tested on the H. sapiens (H_200) dataset and by about 6.5%, 35.0%, and 150.0% when tested on the S. cerevisiae (S_200) dataset, respectively. This significant growth in MCC is very important since it ensures the stability and performance of our model. With those two independent test datasets, our model also presented higher accuracy with a success rate boosted by 7.0%, 13.0%, and 20.0% and 2.0%, 9.5%, and 25.0% when compared to iPseU-CNN, PseUI, and iRNA-PseU, respectively. For the majority of other evaluation metrics, iPseU-NCP demonstrated superior performance as well.
Conclusions: iPseU-NCP combining the RF and NPC-encoded features showed better performances than other existing state-of-the-art methods in the identification of pseudouridine sites. This also shows an optimistic view in addressing biological issues related to human diseases.
Click here for Demo